Nature has published a survey article, “What’s next for AlphaFold and the AI protein-folding revolution”, which discusses the impact upon molecular biology of the release of AlphaFold, an artificial intelligence program developed by DeepMind, a subsidiary of Alphabet/Google, which predicts the three-dimensional structure into which proteins fold from the sequence of nucleotides that encode the amino acids from which the molecule is assembled. This “protein folding problem” was long believed to be computationally intractable, leaving researchers only difficult and time-consuming methods such as X-ray crystallography and nuclear magnetic resonance to tease out the three-dimensional structure of the molecules of biology and figure out how these molecular machines work.
In July of 2021, DeepMind released AlphaFold, a deep learning program for protein folding, to the public. Peter Beck, who experimentally studies protein folding at the Max Planck Institute in Frankfurt, Germany says, “AlphaFold changes the game. This is like an earthquake. You can see it everywhere.” Ora Schueler-Furman of the Hebrew University in Jerusalem said, “There is before July and after.”
Here is a chart showing the AlphaFold effect in terms of the number of citations of AlphaFold2 in scientific publications since December 2020.
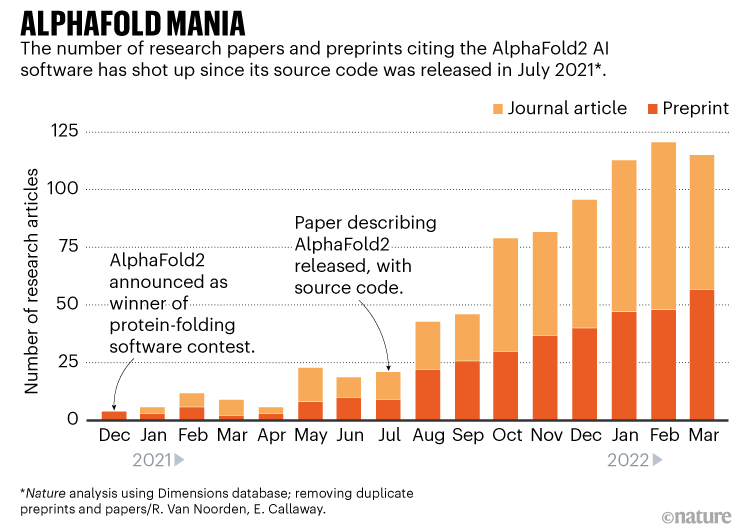
This year, DeepMind plans to release a total of more than 100 million structure predictions. That is nearly half of all known proteins — and hundreds of times more than the number of experimentally determined proteins in the Protein Data Bank (PDB) structure repository.
AlphaFold deploys deep-learning neural networks: computational architectures inspired by the brain’s neural wiring to discern patterns in data. It has been trained on hundreds of thousands of experimentally determined protein structures and sequences in the PDB and other databases. Faced with a new sequence, it first looks for related sequences in databases, which can identify amino acids that have tended to evolve together, suggesting they’re close in 3D space. The structure of existing related proteins provides another way to estimate distances between amino-acid pairs in the new sequence.
⋮
By now, more than 400,000 people have used the EMBL-EBI’s AlphaFold database, according to DeepMind. There are also AlphaFold ‘power users’: researchers who’ve set up the software on their own servers or turned to cloud-based versions of AlphaFold to predict structures not in the EMBL-EBI database, or to dream up new uses for the tool.
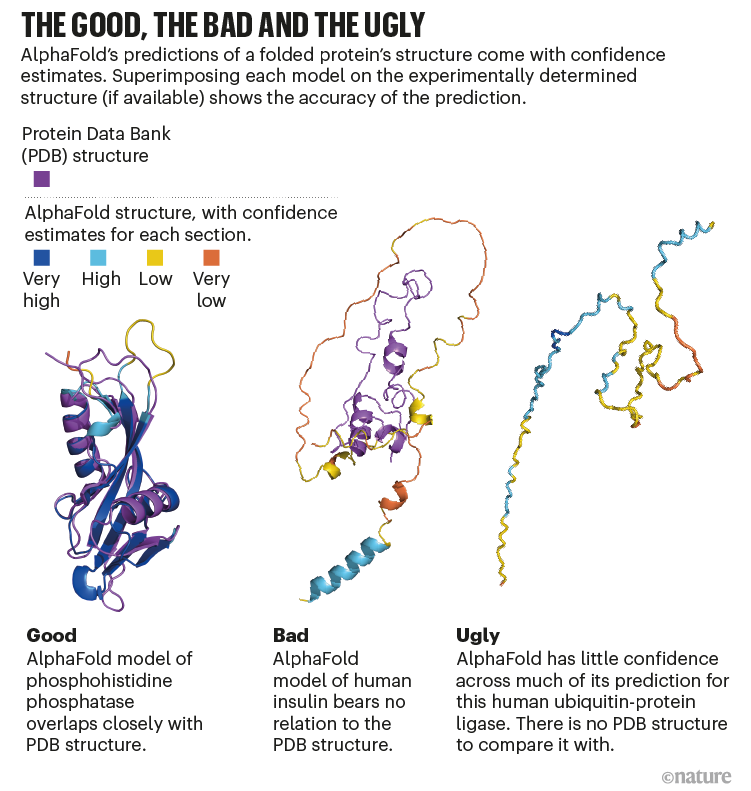
Because AlphaFold works largely by having been trained on known protein structures, it may have difficulties with novel proteins unlike any previously solved. But it is able to assign confidence to the solutions it predicts.
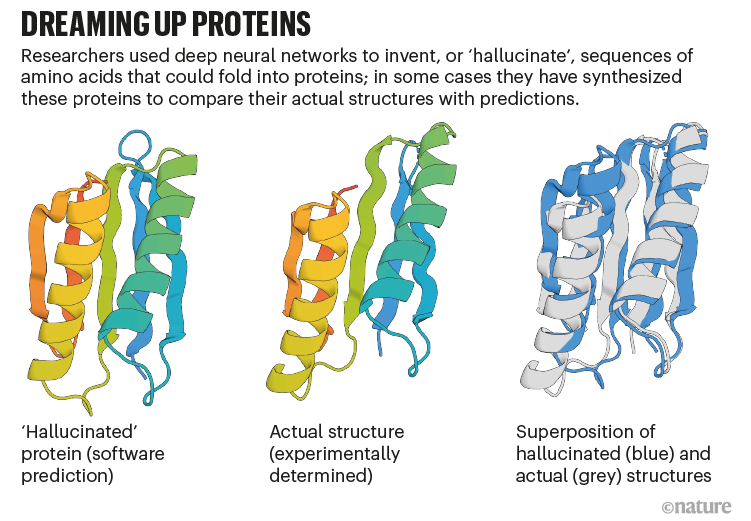
AlphaFold can be used by drug designers and those experimenting with biomimetic nanotechnology to “hallucinate” proteins that do not exist in nature, then study how they will fold when synthesised.
This is a revolution in molecular biology that you can watch while it happens, based upon open source software you can run on your own computer or cloud server, examine how it operates, and investigate improvements to the technologies. Where’s it all going?
Things are just changing so fast,” says [University of Washington in Seattle biochemist David] Baker. “Even in the next year, we’re going to see really major breakthroughs made using these tools.” Janet Thornton, a computational biologist at the EMBL-EBI, thinks one of AlphaFold’s biggest impacts might be simply to convince biologists to be more open to insights from computational and theoretical approaches. “To me, the revolution is the mindset change,” she says.
Here is a video from DeepMind about the development of AlphaFold.
